 打印
打印
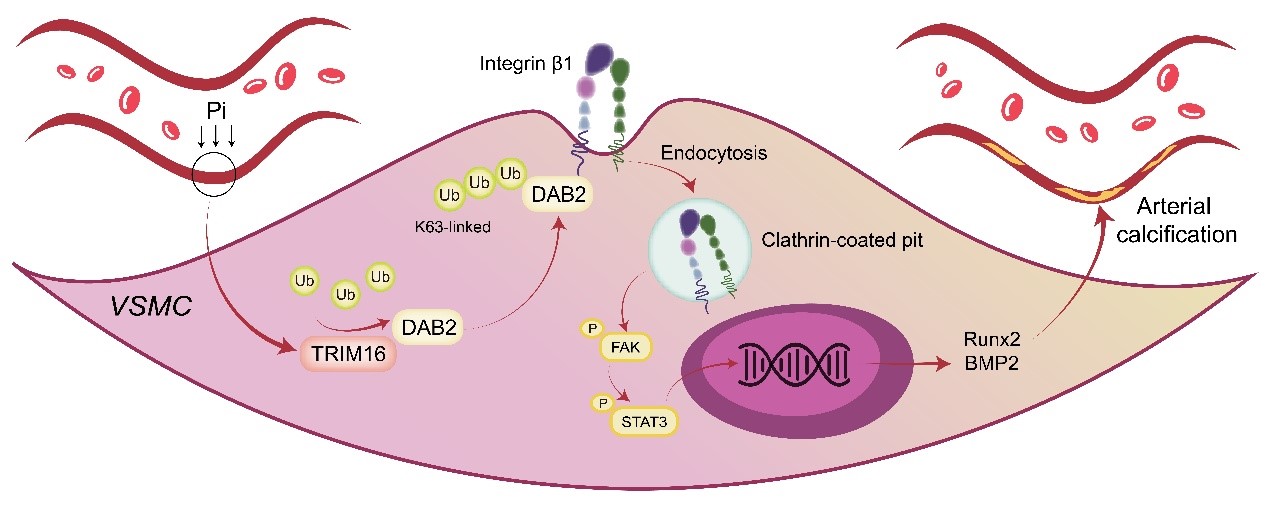
图 TRIM16促进慢性肾病血管钙化机制示意图
在国家自然科学基金项目(批准号:82370418、82270426、82170414)等资助下,南方医科大学颜建云教授联合中山大学陆立鹤教授团队在慢性肾病血管钙化的发病机制研究方面取得进展,研究成果以“TRIM16介导DAB2的K63连接泛素化促进血管钙化(TRIM16 Mediates K63-Linked Ubiquitination of DAB2 to Facilitate Vascular Calcification)”为题,于2025年8月发表于《循环研究》(Circulation Research)杂志,原文链接:https://www.ahajournals.org/doi/full/10.1161/CIRCRESAHA.125.326520。
慢性肾病(chronic kidney disease,CKD)是一种严重威胁人类健康的疾病。血管钙化可引起血管弹性的降低、动脉顺应性下降及心脏负荷加重,是导致CKD患者心血管不良事件发生和死亡的重要危险因素。目前CKD血管钙化的分子机制尚未完全阐明,临床上对于血管钙化尚缺乏有效的治疗手段。
研究团队发现,在多种慢性肾病动物血管钙化模型中三联结构域16(tripartite motif 16,TRIM16)表达显著升高,过表达TRIM16促进了CKD大鼠主动脉钙盐沉积,敲低TRIM16则明显减轻CKD大鼠主动脉钙化;在腺嘌呤饮食诱导的CKD小鼠模型中,TRIM16基因敲除同样可显著延缓主动脉钙化的进展。研究团队通过免疫共沉淀联合质谱的方法,发现失能同源物2(disabled homolog 2,DAB2)是TRIM16的主要结合蛋白,并揭示了TRIM16与DAB2的蛋白互作关系,同时解析了二者特异性结合的结构域。研究者进一步发现,TRIM16通过催化DAB2蛋白的泛素化修饰,进而增强其介导的整合素β1内吞转运,激活下游的FAK-STAT3信号通路,最终促进血管钙化(图)。
该研究明确了TRIM16是调控血管钙化病理进程关键E3泛素连接酶,同时揭示了非蛋白酶体依赖型泛素化修饰参与调控血管钙化的新机制,为CKD血管钙化提供了潜在的治疗靶标。
