 打印
打印
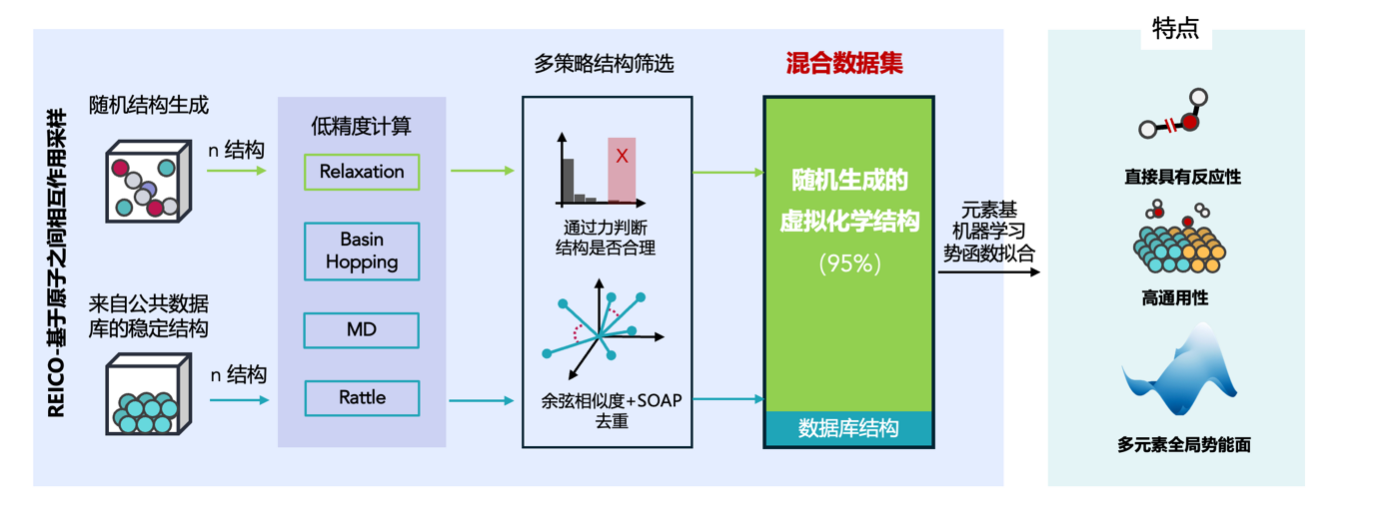
在国家自然科学基金项目(批准号:22433004、22403064)资助下,上海科技大学谢闻博与胡培君团队共同提出了以原子间相互作用为核心的训练集采样方法,并开发了基于元素的通用型机器学习势能训练框架。相关成果以“General reactive element-based machine learning potentials for heterogeneous catalysis”为题,于2025年9月23日在线发表在《Nature Catalysis》上。文章链接:https://www.nature.com/articles/s41929-025-01398-3。
密度泛函理论(DFT)一直是理解催化微观反应机制的有效方法。然而,在真实反应条件下的多相催化机理却极其复杂,尤其受表面覆盖度效应、动态结构演化过程及界面环境等关键因素影响。若这些因素共同耦合将直接导致DFT计算量呈指数级上升。发展机器学习势函数可在一定程度上弥合计算效率与精度的矛盾,但往往受限于数据采样策略。因此,如何同时保证计算效率与精度,发展类似 DFT有效计算方法,已成为理论化学领域的关键科学问题之一。
上述研究团队提出了一种新势函数采样方法,可摆脱对特定结构与反应坐标的依赖,把采样对象从“结构空间”转向“原子相互作用空间”,更加聚焦于局部原子环境的多样性。该新方法可通过随机生成包含虚拟化学结构的“小体系”,在其优化轨迹中捕捉丰富的化学键形成与断裂、配位变化等局部环境,从而构建出具有高度代表性和多样性的训练数据集。重要的是,该方法赋予模型真正的可迁移能力,使之可直接应用于实际大体系,实现了接近 DFT 计算的通用预测,并在多种多相催化体系中验证其有效性。
该研究工作通过单一模型实现了气、固、液等多相体系的统一精确描述,为大规模复杂催化体系模拟提供了高效可靠的DFT计算替代方案,同时将机器学习势函数从面向单一问题拓展至面向多相催化体系的通用计算框架。
