 打印
打印
在国家自然科学基金项目(批准号:32394032)等资助下,中国人民解放军第四军医大学武胜昔教授和王亚周教授团队,在孤独症社交障碍的分子神经机制研究方面取得进展。研究成果以“H2S介导的突触蛋白异常硫巯基化参与孤独症社交障碍(Mitochondrial dysfunction reveals H2S-mediated synaptic sulfhydration as a potential mechanism for autism-associated social defects)”为题,于2025年9月3日发表于《细胞·代谢》(Cell Metabolism)杂志。论文链接:https://doi.org/10.1016/j.cmet.2025.08.003。
社交障碍(social dysfunction)是孤独症最重要的临床症状之一,至今缺乏有效治疗方法。通常认为,社交相关脑区的突触发育或功能异常是孤独症社交障碍的重要原因。由于孤独症是一种病因异质性极高的疾病,不同的基因突变是否在神经元胞内引起某种共性的病理改变,从而导致突触发育/功能异常?这一直是领域内一个悬而未解的重要问题。
针对孤独症社交障碍机制不明且缺乏共性病理机制的挑战,该团队从神经代谢与蛋白修饰调控的角度开展研究,采用两种遗传因素致孤独症小鼠(Shank3-/-和Fmr1-/y小鼠),发现其前扣带回皮质(ACC)存在线粒体功能障碍。代谢组学分析发现该两种小鼠ACC中半胱氨酸代谢异常,H2S水平升高。抑制ACC脑区的H2S合成关键酶CBS,可改善孤独症小鼠的社交功能;团队进一步的通过硫巯基化修饰组学、电生理等多种技术手段,揭示该两种小鼠ACC脑区大量突触蛋白发生过度硫巯基化修饰,突变代谢型谷氨酸受体mGluR5的关键硫巯基化位点可部分挽救该两种孤独症小鼠的社交障碍。在此基础上,团队还开发出一种低硫饲料干预方案,成功改善孤独症模型小鼠的突触功能与社交行为,并在人神经元模型及孤独症患者样本中验证了该机制的保守性,为孤独症社交障碍的治疗提供了新靶点和干预策略(图)。
该研究深化了对孤独症发病机制与治疗策略的认识,从以往主要关注遗传突变与突触蛋白的直接作用,拓展至对“线粒体—小分子代谢—蛋白修饰”这一调控网络的系统解析,开辟了通过代谢干预与修饰调控治疗神经发育障碍的新路径。
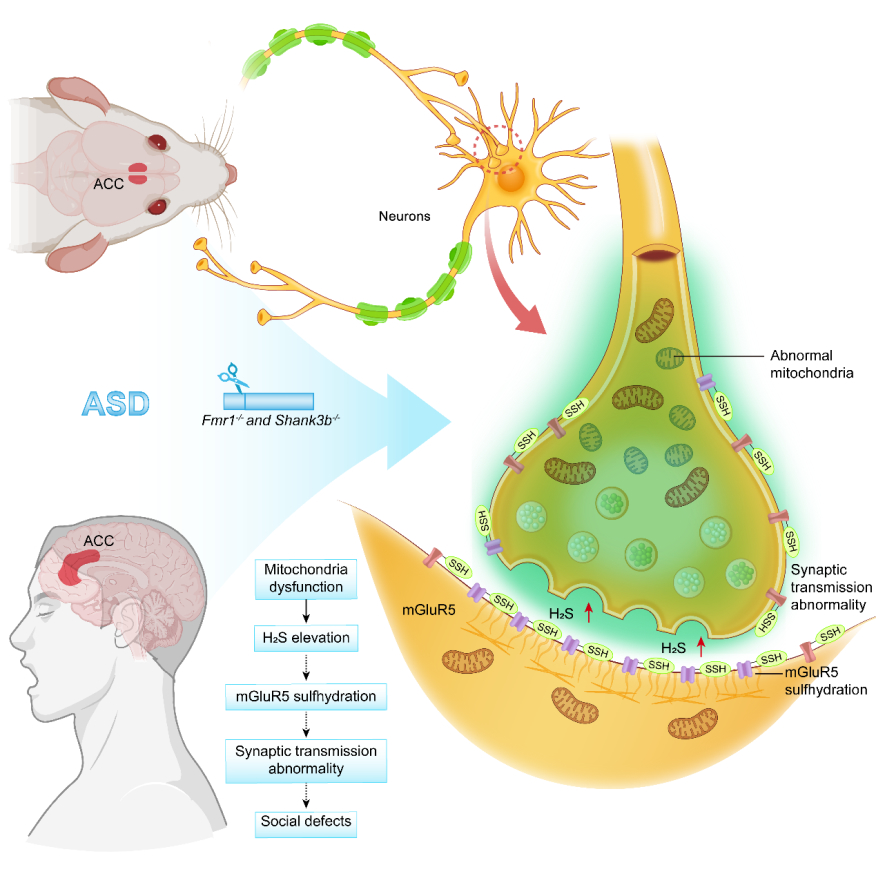
图 前扣带回皮质H2S介导突触蛋白异常硫巯巯基化参与孤独症社交障碍
