 打印
打印
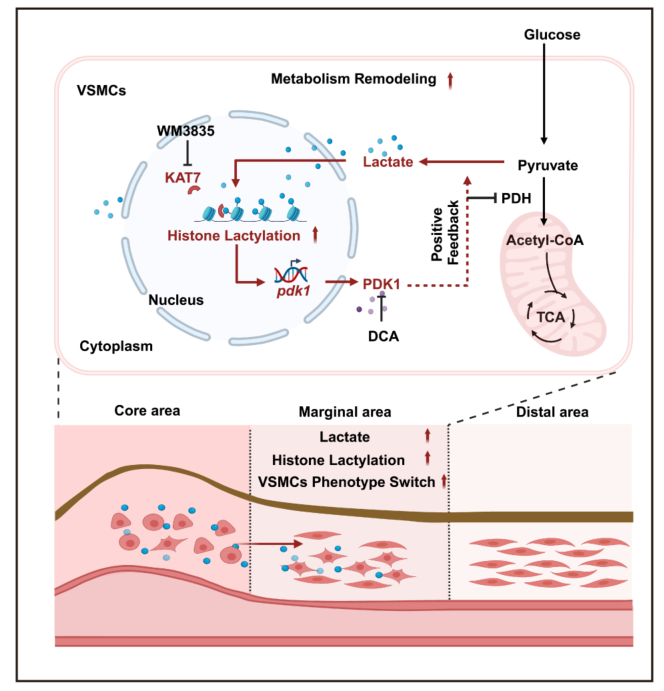
图 组蛋白乳酸化介导的KAT7-H4K16la-PDK1-乳酸正反馈环路驱动主动脉瘤/夹层进展的机制示意图
在国家自然科学基金项目(批准号:T2288101、82500567、82400554等)资助下,复旦大学附属中山医院葛均波院士、孙爱军教授团队在主动脉瘤/夹层发病机制与干预靶点研究方面取得进展。研究成果以“组蛋白乳酸化介导的血管平滑肌细胞代谢重塑通过促进乳酸积累加重主动脉瘤/夹层(Histone lactylation-mediated metabolic remodeling in vascular smooth muscle cells aggravates aortic aneurysm and dissection via promoting lactate accumulation)”为题,于2026年1月5日在《循环》(Circulation)杂志发表。论文链接:https://doi.org/10.1161/CIRCULATIONAHA.125.072576。
主动脉瘤和夹层(AAD)是严重威胁生命的心血管急重症,目前缺乏有效的药物干预手段。血管平滑肌细胞(VSMCs)表型转换伴随代谢重编程是AAD进展的关键驱动因素,但其表观遗传调控机制尚不明确。
该研究发现AAD患者及模型小鼠主动脉组织中组蛋白乳酸化水平显著升高,且特异性发生在VSMCs,其中H4K16la是关键修饰位点。研究团队进而利用血管紧张素II诱导的AAD小鼠和细胞模型,在微观层面揭示H4K16la富集于PDK1启动子区,促进其转录,推动乳酸生成;而积累的乳酸又进一步增强H4K16la修饰,形成正反馈环路,驱动VSMCs代谢重编程和表型转换。该过程由赖氨酸转移酶KAT7催化介导。基于上述发现,团队采用PDK1抑制剂DCA或KAT7抑制剂WM3835靶向干预,显著抑制组蛋白乳酸化,恢复VSMCs收缩表型,减少主动脉扩张,降低小鼠死亡率。进一步临床证据表明,AAD患者血乳酸水平显著升高,且与H4K16la水平正相关,提示二者可作为潜在诊断生物标志物(图)。
该研究不仅揭示了“KAT7-H4K16la-PDK1-乳酸”正反馈环路在AAD进展中的核心作用,为理解血管疾病的代谢-表观遗传机制提供了全新视角,更明确了PDK1和KAT7作为治疗靶点的潜力,同时为AAD的早期诊断提供了新的生物标志物策略。