|
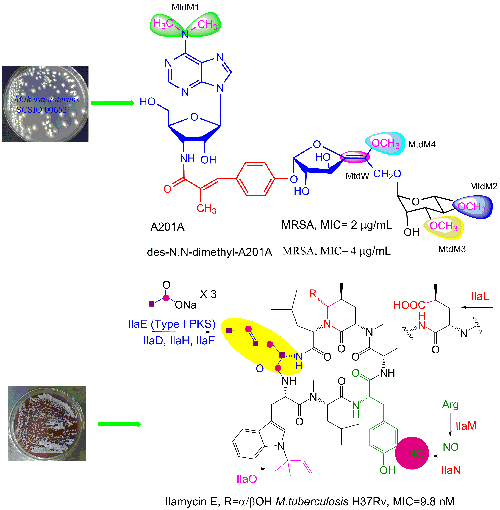 |
图. 抗感染抗生素的发现及其生物合成技术改造
在国家自然科学基金项目(项目编号:81425022,31270134,U1501223)等资助下,中国科学院南海海洋研究所鞠建华研究员团队在抗感染抗生素的发现和生物合成研究方面取得新进展。相关研究结果以“Deciphering the Sugar Biosynthetic Pathway and Tailoring Steps of Nucleoside Antibiotic A201A Unveils a GDP-l-galactose Mutase”(核苷类抗生素A201A的糖基后修饰及其GDP-l-半乳糖变位酶的阐明)和“Biosynthesis of Ilamycins Featuring Unusual Building Blocks and Engineered Production of Enhanced Anti-tuberculosis Agents”(怡莱霉素的生物合成和利用基因工程改造得到强效抗结核抗生素)为题,于近期分别发表在PNAS和Nature Communications(《自然通讯》)上,论文链接:http://www.pnas.org/content/114/19/4948.long和https://www.nature.com/articles/s41467-017-00419-5.pdf。
随着细菌耐药性的发展,抗生素耐药性已成为危害公共卫生安全的重大隐患。为应对这一挑战,发现结构新颖、作用机制独特的新型抗生素成为迫切需求。海洋微生物因生境独特,具备产生新结构活性次级代谢产物的能力,已成为新药研究开发的重要战略新资源。鞠建华研究员团队长期致力于海洋微生物活性次级代谢产物的发现及其生物合成研究,通过利用“深海环境营养匮乏,生活在深海的微生物为了争夺生存空间,会产生抗生素拮抗周边微生物的生长”这一化学生态学原理从深海微生物中筛选抗菌活性物质。经过活性筛选,他们从深海放线菌SCSIO 00652中分离到对G+菌和厌氧性G-菌具显著抗菌活性的含有氨基己糖结构单元的核苷类抗生素A201A,从深海放线菌SCSIO Zh16中分离到具有显著抗结核分枝杆菌活性的结构新颖的怡莱霉素(下图)。
研究人员利用天然产物化学、分子生物学和生物化学等手段阐明了A201A分子中两个糖结构单元的生物合成和后修饰过程,首次发现并阐明了一个负责吡喃半乳糖和呋喃半乳糖互变的变位酶MtdL,并从利用生物合成技术构建的10个衍生物中筛选得到了一个对加氧西林耐药金黄色葡萄球菌(MRSA)的抑制活性为4 mg/mL且具有更好水溶性的衍生物des-N,N-dimethyl-A201A (PNAS, 2017)。研究人员通过对结构新颖的怡莱霉素的生物合成机制的阐明及基因工程改造,得到一个对结核分枝杆菌M. Tuberculosis H37Rv的抑制活性为9.8 nM的新结构衍生物怡莱霉素E,其活性是一线抗结核药物利福平活性的30倍;对正常细胞的毒性较低,在抗结核活性和细胞毒性之间的选择性指数为400-1500,显示出较好的安全性窗口,具有成药潜力(发明专利申请号:201610806737.1,201610805549.7,201610885104.4);更为重要的是通过基因工程改造的方法实现了该化合物在一个基因工程菌株中的高效生产 (Nature Communications, 2017)。研究工作为我国新型海洋药物的进一步开发提供了自主知识产权的化学实体。